Molecular Systems Biology & Medicine
Disease phenotypes are often shaped by disturbance of the complex system our cells represents. Modulation of single components can influence large parts of the system, driving various disease aspects. To get new insights into these dynamic networks of proteins, lipids, nucleic acids and metabolites it is crucial to study components in their functional context. Quantitative measurements of the system as a whole (at least as much as possible) and dissection of complex processes paint a new bigger picture of biology.
Areas of Research

Systems Medicine
We use multi-layered proteomics and other -omics methods to gain molecular knowledge about disease biology. We explore the mechanisms of host-pathogen interactions, neurodegeneration and cancer to lay a foundation for translational research.
Quantitative Proteomics
Our lab has a strong focus on multiple aspects of mass spectrometry based proteomics. We explore protein dynamics, protein-protein interactions and signaling by a wide range of methods. We´re always trying to extend the limits of proteomics and its use for the research commmunity.

Software
Systems biology and medicine heavily rely on computational analysis. During our exploration of different datasets and proteomics methods, we established several helpful scripts, packages and tools, that we share with the research community.
Our Technologies
Besides classical whole cell proteomics, where we measure total protein levels from cells or tissues by Tandem Mass Tags (TMT), we also have palette of more specialized techniques:
Protein Dynamics
This includes measurements of protein translation and degradation. We commonly apply pSILAC in conjunction with Tandem Mass Tags.
Protein-Protein Interactions
We commonly apply TurboID experiments for proximity labelling. Additionally we perform endogenous IPs with antibodies or Affinity purification of tagged protein species.
Modification profiling
We routinely perform phosphorylation analysis and ubiquinitation profiling to dissect signaling networks.
Systems Medicine
Host virus interactions: The infection biology of SARS-CoV-2
In close collaboration with the virology department of the university clinic here in Frankfurt we were able to measure the first quantitative proteomics dataset of human cells infected with SARS-CoV-2. We applied multi-layered proteomics to quantify the dynamic changes of protein synthesis, PTMs and total protein levels over the infection course. We analysed the changes to the host cell proteome to identify pathways important for viral replication. By the identification of these pathways we detected vulnerabilities of the viral replication process that can be targeted by already exisiting drugs. Repurposing these drugs we were able to block the proliferation of the virus in vitro and lay a molecular groundwork for future clincal research.
Bojkova, D., Klann, K., Koch, B. et al. Nature 2020. 583, 469–472
Klann K, Bojkova D et al. Mol Cell 2020. 80 (1) 164-174.e4
Kohli A et al. Life Sci Alliance 2022 Feb 2;5(5):e202201371.
Cancer research: Dissecting proteome dynamics of tumors
 Image: National Cancer Institute
Image: National Cancer Institute
Cancer cells exhibit critical rearrangements of the proteome. The changes of total protein levels, PTM patterns and protein dynamics finally lead to the diverse disease phenotypes. Profiling the alterations on a system-wide scale and on different layers provides an essential groundwork for translational research. The recently founded Frankfurt Cancer Institute partners scientists from a broad spectrum of disciplines, ranging from basic research to all clinical aspects of cancer. Embedded in this great scientific environment we are currently studying the proteome changes during cancer progression to understand disease mechanisms. Therefore, we are applying a wide range of mass-spectrometry powered proteomics ranging from interaction proteomics over PTM profiling to protein dynamics measurements. Tailoring these assays to different models and scientific questions will drive research of many aspects of cancer.
Nguyen et al. Blood 2019, 133(2), 168-179
Koschade SE et al. Leukemia 2022
Quantitative Proteomics
The dynamic proteome: Expanding the toolkit for translation proteomics
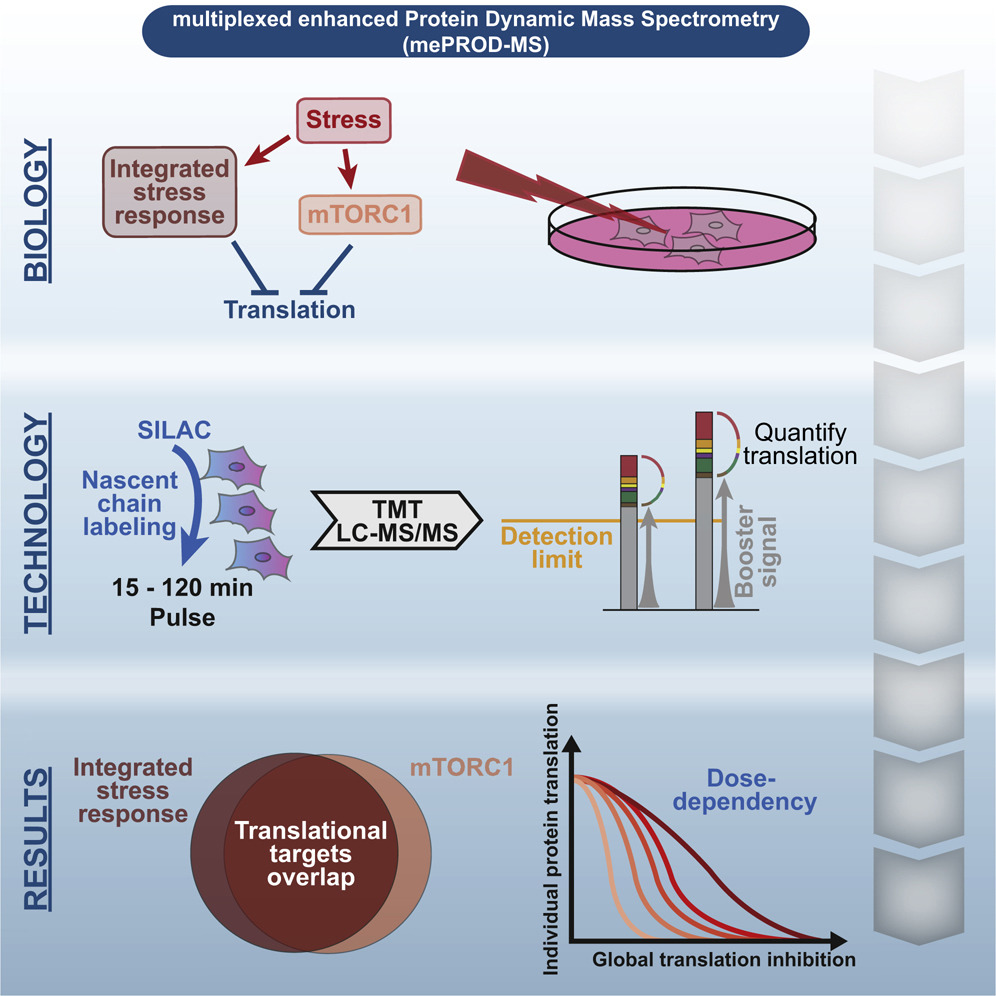
Cells can adapt rapidly to changing environments. These responses are often shaped by alterations in the translational landscape of the cell. Quantifying these changes in an unbiased way provides with new insights to translational regulation and molecular mechanisms. To facilitate faithful translation quantification and to overcome technical limitations we developed multiplexed enhanced Protein Dynamics (mePROD) mass spectrometry. The combination of Tandem Mass Tags and SILAC pulse labeling together with a carrier or booster signal enables us to profile translatome wide changes in the range of minutes without loosing depth of anaylsis. mePROD is able to accurately measure the translation of thousands of proteins, even under conditions where global translation rates are effected (e.g. stress responses). We successfully appied mePROD to diverse disease scenarios, such as viral infection. We currently integrating functional translation proteomics into a wide range of applications to spark new ideas in disease biology.
Klann et al. Molecular Cell 2020, 77(4), 913-925.e4
Klann et al. Analytical Chemistry 2020, 92, 12, 8041-8045
Bojkova, Klann, Koch et al. Nature 2020, 583, 469-472
Schäfer JA, Bozkurt S, Michaelis JB et al. Molecular Cell 2022, 82(2), 435-446
A detailed protocol can be found here:
Klann and Münch, Methods Mol Biol 2022,2428:75-87.
Interaction networks: Profiling protein-protein connections of functional subnetworks
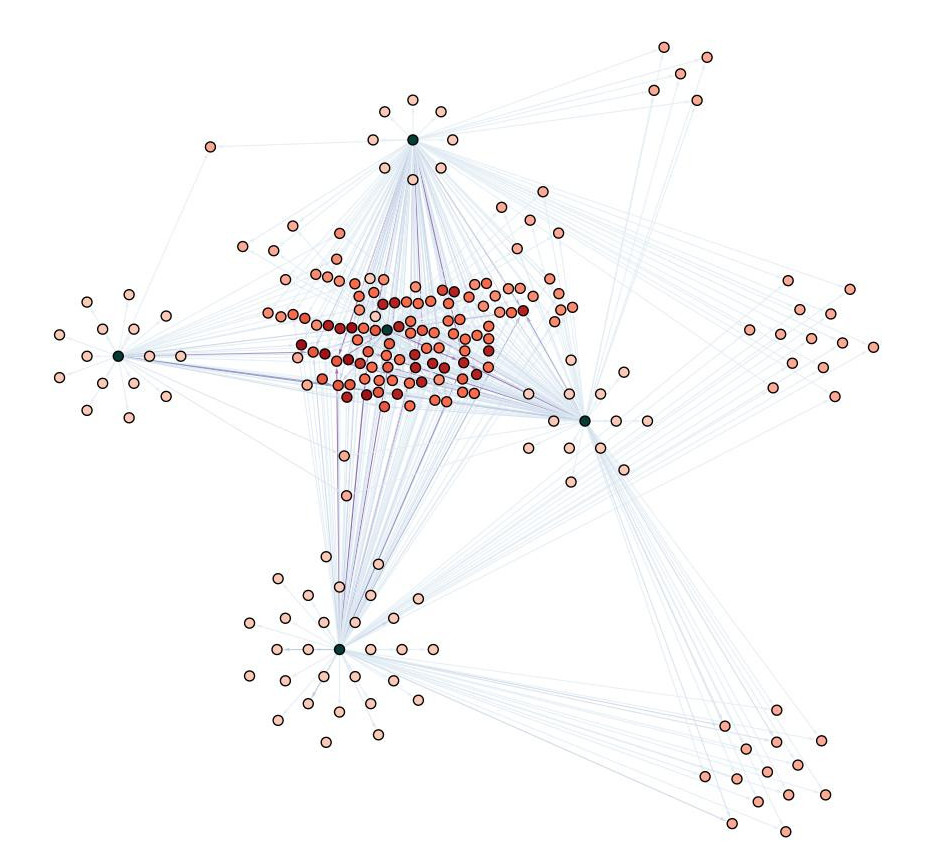
Protein-Protein interactions are essential for cellular function. Upon changing environments proteins can immediately change their interaction partners to fulfill new functions or modulate activities. These changes in activity often cannot be detected at the level of total protein amounts, but contain crucial information about cellular processes. To profile these interaction networks we mainly apply the TurboID system, developed by the Ting lab in Stanford. We are applying this powerful tool to examine how protein-protein interaction networks dynamically change upon stress conditions.
Software
DynaTMT: Analysis of SILAC/TMT data
The emerging role of protein dynamics measurements in the last years, creates the need of sophisticated tools for data analysis. Pulsed SILAC combined with Tandem Mass Tags has evolved as a gold standard for profiling protein synthesis and degradation by mass spectrometry. Additionally the combination of SILAC and TMT to expand the multiplexing capacity of proteomics experiments has been introduced recently. While the experimental setup is available to more researchers, the data analysis still proves difficult. The introduction of two different labels creates a complexity of the data, that needs to be dissected during analysis. DynaTMT is a browser based application (accompanied from a python packge for integration into existing analysis workflows) that extracts the relevant data from mePROD experiments or any combination of SILAC and TMT. The application and the python packages are freely available under the GNU GPLv3 license:
DynaTMT browsertool
DynaTMT python package
DynaTMT publication in Molecular Cell
PBLMM: Peptide-based linear mixed models
Here we present PBLMM, a standalone desktop application for differential expression analysis of proteomics data, together with a python package that allows streamlined data analysis workflows implementing the PBLMM algorithm. PBLMM is easy to use without scripting experience and calculates differential expression by peptide based linear mixed regression models. The application and the python packages are freely available under the GNU GPLv3 license:
PBLMM desktop application
mssuite python package