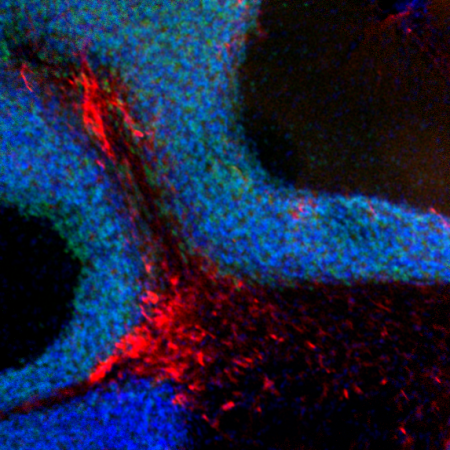
Amyotrophic lateral sclerosis (ALS) is the most common variant of motor neuron disease affecting adults. It is well established that autophagy is important for neuronal homeostasis and that deregulation of autophagy can lead to neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease, as well as ALS. Recent studies have shown that mutations in the CCD2 domain of TBK1 can impair its binding to the autophagy receptor OPTN and cause ALS (Freischmidt et al., 2015). The TBK1-mediated phosphorylation of autophagy receptors, especially OPTN, creates a signal amplification loop operating in selective autophagy of damaged mitochondria (Herhaus et al., 2015). Constitutive interaction of TBK1 with OPTN and the ability of OPTN to bind to ubiquitin chains are essential for TBK1 recruitment and kinase activity during mitophagy. The TBK1-mediated phosphorylation event on OPTN promotes recruitment and retention of OPTN/TBK1 on ubiquitinated, damaged mitochondria (Richter et al., 2016). In addition to phosphorylating autophagy receptors, we recently showed that TBK1 can also phosphorylate ATG8 proteins and thereby influence autophagy flux (Herhaus et al., 2020). Moreover, TBK1 is known to play a role in neuroinflammation, a common symptom of neurodegenerative diseases. Our lab is interested to decipher the molecular mechanisms underlying ALS and other neurodegenerative diseases.
 PROTEOMICS
PROTEOMICS MICROSCOPY
MICROSCOPY TRAFFICKING
TRAFFICKING ER-PHAGY
ER-PHAGY DNA DAMAGE
DNA DAMAGE